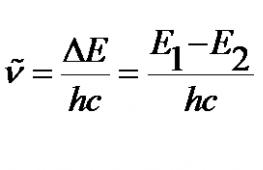Molekulaarspektrite tüübid. Molekulaarspektrid
Molekulaarspektrite uuringud võimaldavad määrata molekulis aatomite vahel mõjuvaid jõude, molekuli dissotsiatsioonienergiat, selle geomeetriat, tuumadevahelisi kaugusi jne. , st. annab ulatuslikku teavet molekuli struktuuri ja omaduste kohta.
Molekulaarspekter viitab laiemas tähenduses üleminekute tõenäosuse jaotusele molekuli kahe energiataseme vahel (vt joonis 9) sõltuvalt üleminekuenergiast. Kuna edaspidi räägime optilistest spektritest, peab iga sellise üleminekuga kaasnema footoni kiirgus või neeldumine energiaga
E n = hn = E 2 – E 1, 3.1
kus E 2 ja E 1 on nende tasandite energiad, mille vahel üleminek toimub.
Kui gaasimolekulide kiirgavatest footonitest koosnev kiirgus lastakse läbi spektraalseadme, siis saadakse molekuli emissioonispekter, mis koosneb üksikutest heledatest (võib-olla värvilistest) joontest. Lisaks vastab iga rida vastavale üleminekule. Omakorda sõltub joone heledus ja asend spektris vastavalt ülemineku tõenäosusest ja footoni energiast (sagedusest, lainepikkusest).
Kui vastupidi, kõigi lainepikkustega footonitest (pidev spekter) koosnev kiirgus lastakse läbi selle gaasi ja seejärel läbi spektraalseadme, siis saadakse neeldumisspekter. Sel juhul on see spekter heleda pideva spektri taustal tumedate joonte kogum. Siirdetõenäosusest ja footoni energiast sõltuvad siin ka joone kontrastsus ja asend spektris.
Lähtudes molekuli energiatasemete keerulisest struktuurist (vt joon. 9) saab kõik nendevahelised üleminekud jagada eraldi tüüpideks, mis annavad molekulide spektrile erineva iseloomu.
Spektrit, mis koosneb joontest, mis vastavad pöörlemistasandite vahelistele üleminekutele (vt joonis 8), muutmata molekuli võnke- ja elektronseisundeid, nimetatakse molekuli pöörlemisspektriks. Kuna pöörleva liikumise energia jääb vahemikku 10 -3 -10 -5 eV, peaks nende spektrite joonte sagedus asuma raadiosageduste mikrolainepiirkonnas (kaug-infrapuna piirkonnas).
Spektrit, mis koosneb joontest, mis vastavad üleminekutele ühes ja samas elektroonilises olekus oleva molekuli erinevatesse võnkeolekutesse kuuluvate pöörlemistasandite vahel, nimetatakse molekuli võnke-rotatsiooni- või lihtsalt võnkespektriks. Need spektrid vibratsioonienergiaga 10 -1 -10 -2 eV asuvad infrapuna sagedusalas.
Lõpuks nimetatakse spektrit, mis koosneb joontest, mis vastavad molekuli erinevatesse elektroonilistesse ja vibratsioonilistesse olekutesse kuuluvate pöörlemistasandite vahelistele üleminekutele, molekuli elektroonika-vibratsiooni-pöörlemis- või lihtsalt elektronspektriks. Need spektrid asuvad nähtava ja ultraviolettkiirguse sageduse piirkonnas, sest elektroonilise liikumise energia on mitu elektronvolti.
Kuna footoni emissioon (või neeldumine) on elektromagnetiline protsess, siis on selle vajalik tingimus molekulis vastava kvantsiirdega seotud elektrilise dipoolmomendi olemasolu või täpsemalt selle muutumine. Siit järeldub, et pöörlemis- ja võnkespektreid saab vaadelda ainult nende molekulide puhul, millel on elektriline dipoolmoment, s.t. mis koosneb erinevatest aatomitest.
Spekter on elektromagnetilise kiirguse energiakvantide jada, mis neeldub, eraldub, hajub või peegeldub aines aatomite ja molekulide üleminekul ühest energiaolekust teise.
Sõltuvalt valguse ja aine vastastikmõju iseloomust võib spektrid jagada neeldumisspektriteks; heitkogused (emissioon); hajumine ja peegeldus.
Uuritavate objektide puhul kasutatakse optilist spektroskoopiat, s.o. spektroskoopia lainepikkuste vahemikus 10 -3 ÷10 -8 m jagatud aatomiteks ja molekulaarseteks.
Aatomi spekter on joonte jada, mille asukoha määrab elektronide ülemineku energia ühelt tasandilt teisele.
Aatomienergia võib esitada translatsioonilise liikumise kineetilise energia ja elektroonilise energia summana:
kus on sagedus, on lainepikkus, on lainearv, on valguse kiirus, on Plancki konstant.
Kuna elektroni energia aatomis on pöördvõrdeline peakvantarvu ruuduga, saab aatomispektris oleva joone võrrandi kirjutada:
 . .
| (4.12) |
Siin  - elektronide energiad kõrgemal ja madalamal tasemel; - Rydbergi konstant;
- elektronide energiad kõrgemal ja madalamal tasemel; - Rydbergi konstant;  - spektriterminid, mis on väljendatud lainearvude ühikutes (m -1, cm -1).
- spektriterminid, mis on väljendatud lainearvude ühikutes (m -1, cm -1).
Kõik aatomispektri jooned koonduvad lühilainepiirkonnas aatomi ionisatsioonienergiaga määratud piirini, mille järel tekib pidev spekter.
Molekuli energia Esimesel hinnangul võib seda pidada translatsiooni-, pöörlemis-, vibratsiooni- ja elektrooniliste energiate summaks:
 | (4.15) |
Enamiku molekulide puhul on see tingimus täidetud. Näiteks H 2 puhul temperatuuril 291 K erinevad koguenergia üksikud komponendid suurusjärgu või rohkem:
309,5 kJ/mol,
 =25,9 kJ/mol,
=25,9 kJ/mol,
2,5 kJ/mol,
 =3,8 kJ/mol.
=3,8 kJ/mol.
Kvantide energiaväärtusi spektri erinevates piirkondades võrreldakse tabelis 4.2.
Tabel 4.2 – Molekulide optilise spektri erinevate piirkondade neeldunud kvantide energia
Mõisted "tuumade vibratsioon" ja "molekulide pöörlemine" on suhtelised. Tegelikkuses annavad sellised liikumistüübid vaid väga ligikaudselt edasi ideid tuumade jaotumisest ruumis, mis on sama tõenäosusliku iseloomuga kui elektronide jaotus.
Skemaatiline energiatasemete süsteem kaheaatomilise molekuli puhul on toodud joonisel 4.1.
Üleminekud pöörlemisenergia tasemete vahel toovad kaasa pöörlemisspektrite ilmnemise kaugemates IR- ja mikrolainepiirkondades. Üleminekud vibratsioonitasemete vahel samal elektroonilisel tasemel annavad võnke-pöörlemisspektrid lähi-IR-piirkonnas, kuna vibratsioonikvantarvu muutus toob paratamatult kaasa pöörlemiskvantarvu muutumise. Lõpuks põhjustavad üleminekud elektrooniliste tasemete vahel elektrooniliste vibratsiooni-pöörlemisspektrite ilmnemist nähtavas ja UV-piirkonnas.
Üldjuhul võib üleminekute arv olla väga suur, kuid tegelikult ei ilmu need kõik spektritesse. Üleminekute arv on piiratud valikureeglid .
Molekulaarspektrid pakuvad palju teavet. Neid saab kasutada:
Ainete tuvastamiseks kvalitatiivses analüüsis, kuna igal ainel on oma ainulaadne spekter;
Kvantitatiivseks analüüsiks;
Struktuurirühmade analüüsiks, kuna teatud rühmad, nagu >C=O, _ NH 2, _ OH jne, annavad spektrites iseloomulikke ribasid;
Molekulide energiaseisundite ja molekulaarkarakteristikute (tuumadevaheline kaugus, inertsimoment, loomulikud võnkesagedused, dissotsiatsioonienergiad) määramiseks; molekulaarspektrite põhjalik uurimine võimaldab teha järeldusi molekulide ruumilise struktuuri kohta;
Kineetilistes uuringutes, sealhulgas väga kiirete reaktsioonide uurimiseks.

 - elektrooniliste loodete energia;
- elektrooniliste loodete energia;
Vibratsioonitasandite energia;
Pöörlemistasandite energiad
Joonis 4.1 – Kaheaatomilise molekuli energiatasemete skemaatiline paigutus
Bouguer-Lambert-Beeri seadus
Molekulaarspektroskoopiat kasutava kvantitatiivse molekulaaranalüüsi aluseks on Bouguer-Lambert-Beeri seadus , mis ühendab langeva ja läbiva valguse intensiivsuse neelava kihi kontsentratsiooni ja paksusega (joonis 4.2):
või proportsionaalsusteguriga:
Integratsiooni tulemus:
 | (4.19) |
 . .
| (4.20) |
Kui langeva valguse intensiivsus väheneb suurusjärgu võrra
 . .
| (4.21) |
Kui =1 mol/l, siis, s.o. Neeldumistegur on võrdne kihi vastastikuse paksusega, milles 1-ga võrdse kontsentratsiooni korral langeva valguse intensiivsus väheneb suurusjärgu võrra.
Neeldumiskoefitsiendid ja sõltuvad lainepikkusest. Selle sõltuvuse tüüp on molekulide "sõrmejälg", mida kasutatakse kvalitatiivses analüüsis aine tuvastamiseks. See sõltuvus on konkreetse aine jaoks iseloomulik ja individuaalne ning peegeldab molekulis sisalduvaid iseloomulikke rühmi ja sidemeid.
Optiline tihedus D
väljendatuna %
4.2.3 Kaheaatomilise molekuli pöörlemisenergia jäiga rotaatori lähenduses. Molekulide pöörlemisspektrid ja nende rakendamine molekulaarkarakteristikute määramiseks
Pöörlemisspektrite tekkimine on tingitud sellest, et molekuli pöörlemisenergia on kvantiseeritud, s.o.
| 0 |
| A |
Alates punktist O on molekuli raskuskese, siis:
Vähendatud massi tähistuse kasutuselevõtt:
 | (4.34) |
viib võrrandini
 . .
| (4.35) |
Seega on kaheaatomiline molekul (joonis 4.7 A), mis pöörleb ümber telje või läbib raskuskeskme, võib lihtsustatult pidada osakeseks massiga , mis kirjeldab ringi raadiusega ümber punkti O(Joonis 4.7 b).
Molekuli pöörlemine ümber telje annab inertsmomendi, mis on praktiliselt võrdne nulliga, kuna aatomite raadiused on palju väiksemad kui tuumadevaheline kaugus. Molekuli sidejoonega vastastikku risti või telgede ümber pöörlemine põhjustab võrdse suurusega inertsimomente:
kus on pöörlemiskvantarv, mis võtab ainult täisarvud
0, 1, 2… Vastavalt pöörlemisspektri valiku reegel kaheaatomilisest molekulist on pöörlemiskvantarvu muutus energiakvanti neelamisel võimalik ainult ühe võrra, s.o.
teisendab võrrandi (4.37) järgmisele kujule:
 20
20  12
6
2
12
6
2 |
joone lainearv pöörlemisspektris, mis vastab kvanti neeldumisele üleminekul j energiatase taseme kohta j+1, saab arvutada võrrandi abil:
Seega on jäiga rotaatori mudeli lähenduses pöörlemisspekter joonte süsteem, mis paiknevad üksteisest samal kaugusel (joonis 4.5b). Jäiga rotaatori mudelis hinnatud kaheaatomiliste molekulide pöörlemisspektrite näited on toodud joonisel 4.6.
| A b |


Joonis 4.6 – Pöörlemisspektrid HF (A) Ja CO(b)
Vesinikhalogeniidi molekulide puhul nihutatakse see spekter spektri kaugemasse IR piirkonda, raskemate molekulide puhul - mikrolaineahju.
Saadud kaheaatomilise molekuli pöörlemisspektri välimuse mustrite põhjal määratakse praktikas kõigepealt kindlaks spektri külgnevate joonte vaheline kaugus, millest need seejärel leitakse, ja kasutades võrrandeid:
 , ,
| (4.45) |
Kus - tsentrifugaalmoonutuse konstant
, on seotud pöörlemiskonstandiga ligikaudse seosega  . Parandust tuleks arvesse võtta ainult väga suurte puhul j.
. Parandust tuleks arvesse võtta ainult väga suurte puhul j.
Polüaatomiliste molekulide puhul on üldiselt võimalikud kolm erinevat inertsimomenti  . Kui molekulis on sümmeetriaelemendid, võivad inertsmomendid langeda kokku või olla isegi võrdsed nulliga. Näiteks lineaarsete polüatomiliste molekulide jaoks(CO 2, OCS, HCN jne)
. Kui molekulis on sümmeetriaelemendid, võivad inertsmomendid langeda kokku või olla isegi võrdsed nulliga. Näiteks lineaarsete polüatomiliste molekulide jaoks(CO 2, OCS, HCN jne)
Kus  - pöörlemisüleminekule vastava joone asukoht
- pöörlemisüleminekule vastava joone asukoht  isotoop-asendatud molekulis.
isotoop-asendatud molekulis.
Joone isotoopnihke suuruse arvutamiseks on vaja järjestikku arvutada isotoopasendatud molekuli vähendatud mass, võttes arvesse isotoobi aatommassi muutust, inertsimomenti, pöörlemiskonstanti ja asendit. joont molekuli spektris vastavalt võrranditele (4.34), (4.35), (4.39) ja (4.43) või hinnata joonte lainearvude suhet, mis vastavad samale üleminekule isotoopasendatud ja mitte -isotoopasendatud molekulid ja seejärel määrake võrrandi (4.50) abil isotoopide nihke suund ja suurus. Kui tuumadevahelist kaugust pidada ligikaudu konstantseks  , siis vastab lainearvude suhe vähendatud masside pöördsuhtele:
, siis vastab lainearvude suhe vähendatud masside pöördsuhtele:
kus on osakeste koguarv, on osakeste arv ühe kohta i- see energiatase temperatuuril T, k- Boltzmanni konstant, - statistiline ve jõudu degeneratsiooni aste i-selle energiatasemega, iseloomustab osakeste leidmise tõenäosust antud tasemel.
Pöörleva oleku korral iseloomustab tasemepopulatsiooni tavaliselt osakeste arvu suhe j- see energiatase nulltasemel olevate osakeste arvu suhtes:
 , ,
| (4.53) |
Kus  - statistiline kaal j-selle pöörlemisenergia tasemega, vastab pöörleva molekuli impulsi projektsioonide arvule selle telje suhtes - molekuli sideliinile,
- statistiline kaal j-selle pöörlemisenergia tasemega, vastab pöörleva molekuli impulsi projektsioonide arvule selle telje suhtes - molekuli sideliinile,  , null pöörlemistasandi energia
, null pöörlemistasandi energia  . Funktsioon läbib maksimumi, kui see suureneb j, nagu on näidatud joonisel 4.7, kasutades näitena CO molekuli.
. Funktsioon läbib maksimumi, kui see suureneb j, nagu on näidatud joonisel 4.7, kasutades näitena CO molekuli.
Funktsiooni ekstreemum vastab tasemele maksimaalse suhtelise populatsiooniga, mille kvantarvu väärtuse saab arvutada võrrandi abil, mis saadakse pärast funktsiooni tuletise määramist ekstreemumis:
 . .
| (4.54) |


Joonis 4.7 – Pöörlemisenergia tasemete suhteline populatsioon
molekulid CO temperatuuridel 298 ja 1000 K
Näide. Pöörlemisspektris HI määratakse külgnevate joonte vaheline kaugus  cm -1. Arvutage molekuli pöörlemiskonstant, inertsimoment ja tasakaaluline tuumadevaheline kaugus.
cm -1. Arvutage molekuli pöörlemiskonstant, inertsimoment ja tasakaaluline tuumadevaheline kaugus.
Lahendus
Jäiga rotaatori mudeli lähenduses määrame vastavalt võrrandile (4.45) pöörlemiskonstandi:
 cm -1.
cm -1.
Molekuli inertsimoment arvutatakse pöörlemiskonstandi väärtusest võrrandi (4.46) abil:
 kg . m 2.
kg . m 2.
Tuumadevahelise tasakaalukauguse määramiseks kasutame võrrandit (4.47), võttes arvesse, et vesiniku tuumade massid  ja jood
ja jood  väljendatud kilogrammides:
väljendatud kilogrammides:
Näide. 1H35Cl spektri kaugemas IR piirkonnas tuvastati read, mille lainenumbrid on:
Määrake molekuli inertsimomendi ja tuumadevahelise kauguse keskmised väärtused. Omistage spektris vaadeldavad jooned pöörlemisüleminekutele.
Lahendus
Jäiga rotaatori mudeli järgi on pöörlemisspektri külgnevate joonte lainearvude erinevus konstantne ja võrdne 2-ga. Määrame pöörlemiskonstandi spektri naaberjoonte vahekauguste keskmisest väärtusest:
![]() cm-1,
cm-1,
 cm -1
cm -1
Leiame molekuli inertsimomendi (võrrand (4.46)):
Arvutame tuumadevahelise tasakaalukauguse (võrrand (4.47)), võttes arvesse, et vesiniku tuumade massid  ja kloor
ja kloor  (väljendatud kilogrammides):
(väljendatud kilogrammides):
Kasutades võrrandit (4.43), hindame joonte asukohta 1 H 35 Cl pöörlemisspektris:
Võrrelgem joonte lainearvude arvutatud väärtusi eksperimentaalsete väärtustega. Selgub, et 1 H 35 Cl pöörlemisspektris täheldatud jooned vastavad üleminekutele:
| N rida | |||||||
| , cm -1 | 85.384 | 106.730 | 128.076 | 149.422 | 170.768 | 192.114 | 213.466 |
 | 3 4 | 4 5 | 5 6 | 6 7 | 7 8 | 8 9 | 9 10 |
Näide. Määrake neeldumisjoone isotoopnihke suurus ja suund, mis vastab üleminekule -ga  energiatase 1 H 35 Cl molekuli pöörlemisspektris, kui kloori aatom asendatakse 37 Cl isotoobiga. Tuumadevahelist kaugust 1 H 35 Cl ja 1 H 37 Cl molekulides peetakse samaks.
energiatase 1 H 35 Cl molekuli pöörlemisspektris, kui kloori aatom asendatakse 37 Cl isotoobiga. Tuumadevahelist kaugust 1 H 35 Cl ja 1 H 37 Cl molekulides peetakse samaks.
Lahendus
Üleminekule vastava joone isotoopnihke suuruse määramiseks , arvutame 1 H 37 Cl molekuli vähendatud massi, võttes arvesse 37 Cl aatommassi muutust:
Järgmiseks arvutame välja inertsmomendi, pöörlemiskonstandi ja joone asukoha  1 H 37 Cl molekuli spektris ja isotoobi nihke väärtus vastavalt võrranditele (4.35), (4.39), (4.43) ja (4.50).
1 H 37 Cl molekuli spektris ja isotoobi nihke väärtus vastavalt võrranditele (4.35), (4.39), (4.43) ja (4.50).
Vastasel juhul saab isotoopnihet hinnata samale üleminekule molekulides vastavate joonte lainearvude suhte järgi (eeldame tuumadevahelise kauguse konstantseks) ja seejärel võrrandi (4.51) abil joone asukoha spektris.
Molekulide 1 H 35 Cl ja 1 H 37 Cl puhul on antud ülemineku lainearvude suhe võrdne:

Isotoopiliselt asendatud molekuli joone lainenumbri määramiseks asendame eelmises näites leitud üleminekulaine numbri väärtusega j → j+1 (3→4):
Me järeldame: isotoopne nihe madala sagedusega või pika laine piirkonda on
85,384-83,049=2,335 cm -1.
Näide. Arvutage 1 H 35 Cl molekuli pöörlemisspektri kõige intensiivsema spektrijoone lainearv ja lainepikkus. Sobitage joon vastava pöördsiirdega.
Lahendus
Molekuli pöörlemisspektri kõige intensiivsem joon on seotud pöörlemisenergia taseme maksimaalse suhtelise populatsiooniga.
Eelmises näites leitud pöörlemiskonstandi väärtuse asendamine 1 H 35 Cl (  cm -1) võrrandisse (4.54) võimaldab meil arvutada selle energiataseme arvu:
cm -1) võrrandisse (4.54) võimaldab meil arvutada selle energiataseme arvu:
 .
.
Sellelt tasemelt pöörleva ülemineku lainenumber arvutatakse võrrandi (4.43) abil:
Leiame võrrandist (4.11) saadud üleminekulainepikkuse, mis on teisendatud järgmise suhtes:
4.2.4 Mitme muutujaga ülesanne nr 11 “Diaatomiliste molekulide pöörlemisspektrid”
1. Kirjutage kvantmehaaniline võrrand, et arvutada kaheaatomilise molekuli kui jäiga rotaatori pöörlemisliikumise energia.
2. Tuletage võrrand kaheaatomilise molekuli kui jäiga pöörleja pöörlemisenergia muutuse arvutamiseks selle üleminekul külgnevale kõrgemale kvanttasemele  .
.
3. Tuletage võrrand kaheaatomilise molekuli neeldumisspektris olevate pöörlemisjoonte lainearvu sõltuvuse kohta pöörlemiskvantarvust.
4. Tuletage võrrand kaheaatomilise molekuli pöörlemise neeldumisspektri naaberjoonte lainearvude erinevuse arvutamiseks.
5. Arvutage kaheaatomilise molekuli pöörlemiskonstant (cm -1 ja m -1) A molekuli pöörlemisneeldumisspektri pikalainelise infrapunapiirkonna kahe kõrvuti asetseva joone lainearvude järgi (vt tabel 4.3).
6. Määrake molekuli pöörlemisenergia A esimesel viiel kvantrotatsiooni tasemel (J).
7. Joonistage skemaatiliselt kaheaatomilise molekuli kui jäiga rotaatori pöörlemisliikumise energiatasemed.
8. Joonistage sellele diagrammile punktiirjoonega molekuli pöörlemiskvanttasemed, mis ei ole jäik rotaator.
9. Tuletage võrrand tuumadevahelise tasakaalukauguse arvutamiseks naaberjoonte lainearvude erinevuse põhjal pöörlemise neeldumisspektris.
10. Määrake kaheaatomilise molekuli inertsimoment (kg. m2) A.
11. Arvutage molekuli vähendatud mass (kg). A.
12. Arvutage molekuli tuumadevaheline tasakaalukaugus (). A. Võrrelge saadud väärtust võrdlusandmetega.
13. Omistage vaadeldud jooned molekuli pöörlemisspektris A rotatsiooniüleminekutele.
14. Arvutage nivoo pöörlemise üleminekule vastava spektrijoone lainearv j molekuli jaoks A(vt tabel 4.3).
15. Arvutage isotoopasendatud molekuli vähendatud mass (kg). B.
16. Arvutage nivoo pöörlemise üleminekuga seotud spektrijoone lainenumber j molekuli jaoks B(vt tabel 4.3). Tuumadevahelised kaugused molekulides A Ja B pidada võrdseks.
17. Määrata isotoopide nihke suurus ja suund molekulide pöörlemisspektris A Ja B pöörlemistasandi üleminekule vastava spektrijoone jaoks j.
18. Selgitage neeldumisjoonte intensiivsuse mittemonotoonse muutumise põhjust molekuli pöörlemisenergia suurenemisel
19. Määrake suurimale suhtelisele populatsioonile vastava pöörlemistasandi kvantarv. Arvutage molekulide pöörlemisspektri kõige intensiivsemate spektrijoonte lainepikkused A Ja B.
Keemilised sidemed ja molekulaarstruktuur.
Molekul - aine väikseim osake, mis koosneb identsetest või erinevatest üksteisega ühendatud aatomitest keemilised sidemed ja on selle põhiliste keemiliste ja füüsikaliste omaduste kandja. Keemilised sidemed tekivad aatomite väliste valentselektronide vastasmõjul. Molekulides leidub kõige sagedamini kahte tüüpi sidemeid: iooniline ja kovalentne.
Iooniline side (näiteks molekulides NaCl, KBr) toimub aatomite elektrostaatilisel vastasmõjul elektroni üleminekul ühelt aatomilt teisele, s.o. positiivsete ja negatiivsete ioonide moodustumise ajal.
Kovalentne side (näiteks H 2 , C 2 , CO molekulides) tekib siis, kui valentselektronid on jagatud kahe naaberaatomi vahel (valentselektronide spinnid peavad olema antiparalleelsed). Kovalentset sidet selgitatakse identsete osakeste, näiteks vesiniku molekulis olevate elektronide eristamatuse põhimõtte alusel. Osakeste eristamatus toob kaasa vahetada interaktsiooni.
Molekul on kvantsüsteem; seda kirjeldab Schrödingeri võrrand, mis võtab arvesse elektronide liikumist molekulis, molekuli aatomite vibratsioone ja molekuli pöörlemist. Selle võrrandi lahendamine on väga keeruline ülesanne, mis tavaliselt jaguneb kaheks: elektronide ja tuumade jaoks. Eraldatud molekuli energia:
kus on elektronide liikumise energia tuumade suhtes, on tuuma vibratsioonide energia (mille tulemusena tuumade suhteline asend perioodiliselt muutub) ja on tuuma pöörlemise energia (mille tulemusena on ruumis olev molekul muutub perioodiliselt). Valem (13.1) ei võta arvesse molekuli massikeskme translatsioonilise liikumise energiat ja molekulis olevate aatomite tuumade energiat. Neist esimest ei kvantitata, seega ei saa selle muutused kaasa tuua molekulaarspektri tekkimist ja teist võib ignoreerida, kui spektrijoonte ülipeent struktuuri ei arvestata. On tõestatud, et eV, ![]() eV,
eV, ![]() eV, seega >>>>.
eV, seega >>>>.
Kõik avaldises (13.1) sisalduvad energiad on kvantiseeritud (see vastab diskreetsete energiatasemete komplektile) ja määratakse kvantarvudega. Üleminekul ühest energiaolekust teise neeldub või eraldub energia D E=hv. Selliste üleminekute käigus muutuvad samaaegselt elektronide liikumise energia, vibratsiooni ja pöörlemise energia. Teooriast ja eksperimendist järeldub, et pöörlemisenergia tasemete D vaheline kaugus on palju väiksem kui vibratsioonitasemete D vaheline kaugus, mis omakorda on väiksem kui kaugus elektrooniliste tasandite vahel D. Joonis 13.1 näitab skemaatiliselt kaheaatomilise energiatasemeid. molekul (näiteks võetakse arvesse ainult kahte elektroonilist taset – näidatud paksude joontega).
 Molekulide struktuur ja nende energiatasemete omadused avalduvad selles molekulaarsed spektrid–
emissiooni (absorptsiooni) spektrid, mis tekivad molekulide energiatasemete vahel toimuvate kvantüleminekute käigus. Molekuli emissioonispektri määrab selle energiatasemete struktuur ja vastavad valikureeglid.
Molekulide struktuur ja nende energiatasemete omadused avalduvad selles molekulaarsed spektrid–
emissiooni (absorptsiooni) spektrid, mis tekivad molekulide energiatasemete vahel toimuvate kvantüleminekute käigus. Molekuli emissioonispektri määrab selle energiatasemete struktuur ja vastavad valikureeglid.
Seega tekivad erinevat tüüpi tasanditevahelised üleminekud erinevat tüüpi molekulaarspektrid. Molekulide kiirgavate spektrijoonte sagedused võivad vastata üleminekutele ühelt elektrooniliselt tasemelt teisele (elektroonilised spektrid) või ühelt vibratsiooni (pöörlemis) tasemelt teisele ( vibratsiooni (rotatsiooni) spektrid).Lisaks on võimalikud ka üleminekud samade väärtustega Ja tasemetele, millel on kõigi kolme komponendi erinevad väärtused, mille tulemuseks on elektrooniline-vibratsiooni- ja vibratsiooni-pöörlemisspekter.
Tüüpilised molekulaarspektrid on triibulised, esindades ultraviolett-, nähtava- ja infrapunapiirkonnas rohkem või vähem kitsaid ribasid.
Kõrge eraldusvõimega spektraalinstrumente kasutades on näha, et ribad on nii tihedalt asetsevad jooned, et neid on raske lahendada. Molekulaarspektrite struktuur on erinevate molekulide puhul erinev ja muutub komplekssemaks, kui aatomite arv molekulis suureneb (täheldatakse ainult pidevaid lairibasid). Ainult polüaatomilistel molekulidel on võnke- ja pöörlemisspektrid, kaheaatomilistel aga need puuduvad. See on seletatav asjaoluga, et kaheaatomilistel molekulidel ei ole dipoolmomente (võnke- ja pöörlemissiirete ajal ei muutu dipoolmoment, mis on vajalik tingimus, et ülemineku tõenäosus erineks nullist). Molekulaarspektreid kasutatakse molekulide struktuuri ja omaduste uurimiseks, neid kasutatakse molekulaarspektraalanalüüsis, laserspektroskoopias, kvantelektroonikas jm.
molekulaarsed spektrid, optilise emissiooni ja neeldumise spektrid, samuti Ramani hajumine, kuuluvad vabad või lõdvalt ühendatud molekulid. M. s. neil on keeruline struktuur. Tüüpiline M. s. - triibulised, neid täheldatakse emissioonis ja neeldumises ning Ramani hajumises enam-vähem kitsaste ribade kujul ultraviolett-, nähtava- ja lähiinfrapuna-aladel, mis lagunevad kasutatavate spektriinstrumentide piisava lahutusvõimega tihedalt asetsevate joonte komplekt. M. s. spetsiifiline struktuur. on erinevate molekulide puhul erinev ja üldiselt muutub see keerulisemaks, kui aatomite arv molekulis suureneb. Väga keeruliste molekulide puhul koosnevad nähtav ja ultraviolettspekter mõnest laiast pidevast ribast; selliste molekulide spektrid on üksteisega sarnased.
M. s. tekkida millal kvantsiirded vahel energiatasemed E' Ja E'' molekulid vastavalt suhtele
h n= E‘ - E‘’, (1)
Kus h n - emiteeritud neeldunud energia footon sagedus n ( h -Plancki konstant ). Ramani hajumisega h n on võrdne langevate ja hajutatud footonite energiate vahega. M. s. palju keerulisem kui joonaatomi spektrid, mille määrab molekuli siseliikumise suurem keerukus kui aatomites. Koos elektronide liikumisega molekulides kahe või enama tuuma suhtes toimub tuumade vibratsiooniline liikumine (koos neid ümbritsevate siseelektronidega) ümber tasakaaluasendi ja molekuli kui terviku pöörleva liikumise. Need kolm liikumistüüpi – elektrooniline, vibratsiooniline ja pöörlev – vastavad kolme tüüpi energiatasemetele ja kolmele spektritüübile.
Kvantmehaanika järgi võib igat tüüpi liikumise energia molekulis võtta ainult teatud väärtusi, s.t see on kvantiseeritud. Molekuli koguenergia E võib ligikaudu esitada selle kolme tüüpi liikumise kvantiseeritud energia väärtuste summana:
E = E email + E loe + E pöörata (2)
Suurusjärgu järgi
Kus m on elektroni mass ja suurus M omab molekulis olevate aatomituumade massi suurusjärku, s.o. m/M~ 10 -3 -10 -5, seega:
E email >> E loe >> E pöörata (4)
Tavaliselt E el umbes mitu ev(mitusada kJ/mol), E loe ~ 10 -2 -10 -1 eV, E pöörlemine ~ 10 -5 -10 -3 ev.
Vastavalt punktile (4) iseloomustab molekuli energiatasemete süsteemi üksteisest kaugel asuvate elektrooniliste tasemete kogum (erinevad väärtused E el at E arv = E pöörlemine = 0), vibratsioonitasemed asuvad üksteisele palju lähemal (erinevad väärtused E ette arvutada E l ja E rotatsioon = 0) ja veelgi tihedamalt asetsevad pöörlemistasemed (erinevad väärtused E pöörlemine etteantud juures E el ja E loendama).
Elektroonilised energiatasemed ( E el punktis (2) vastavad molekuli tasakaalukonfiguratsioonidele (kaheaatomilise molekuli puhul, mida iseloomustab tasakaaluväärtus r 0 tuumadevaheline kaugus r. Iga elektrooniline olek vastab teatud tasakaalukonfiguratsioonile ja teatud väärtusele E el; madalaim väärtus vastab põhienergiatasemele.
Molekuli elektrooniliste olekute komplekti määravad selle elektronkihi omadused. Põhimõtteliselt väärtused E el saab arvutada meetodite abil kvantkeemia, seda probleemi saab aga lahendada ainult ligikaudsete meetoditega ja suhteliselt lihtsate molekulide puhul. Kõige olulisem teave molekuli elektrooniliste tasemete kohta (elektrooniliste energiatasemete asukoht ja nende omadused), mille määrab selle keemiline struktuur, saadakse selle molekulaarstruktuuri uurimisel.
Antud elektroonilise energiataseme väga oluline omadus on väärtus kvantarv S, mis iseloomustab molekuli kõigi elektronide kogupöörlemismomendi absoluutväärtust. Keemiliselt stabiilsetes molekulides on tavaliselt paarisarv elektrone ja nende jaoks S= 0, 1, 2... (elektroonilise põhitaseme jaoks on tüüpiline väärtus S= 0 ja põnevil - S= 0 ja S= 1). Tase koos S= 0 nimetatakse singletideks, kusjuures S= 1 - triplett (kuna interaktsioon molekulis viib nende jagunemiseni c = 2-ks S+ 1 = 3 alamtaset) . KOOS vabad radikaalid neil on nende jaoks reeglina paaritu arv elektrone S= 1 / 2, 3 / 2, ... ja väärtus on tüüpiline nii põhi- kui ka põnevustasemele S= 1/2 (topelttasemed jagunevad c = 2 alamtasandiks).
Molekulide puhul, mille tasakaalukonfiguratsioon on sümmeetriline, saab elektroonilisi tasemeid täiendavalt klassifitseerida. Kahe- ja lineaarsete kolmeaatomiliste molekulide puhul, mille sümmeetriatelg (lõpmatus järjekorras) läbib kõigi aatomite tuumasid , elektroonilisi tasemeid iseloomustavad kvantarvu l väärtused, mis määrab kõigi elektronide kogu orbitaalimpulsi projektsiooni absoluutväärtuse molekuli teljele. Tasemed, mille l = 0, 1, 2, ... on tähistatud vastavalt S, P, D... ja c väärtust näitab vasakus ülanurgas olev indeks (näiteks 3 S, 2 p, ...). Sümmeetriakeskmega molekulide jaoks, näiteks CO 2 ja C 6 H 6 , kõik elektroonilised tasemed on jagatud paaris- ja paarituteks, mis on tähistatud indeksitega g Ja u(olenevalt sellest, kas lainefunktsioon säilitab sümmeetriakeskmes ümberpööramisel oma märgi või muudab seda).
Vibratsioonienergia tasemed (väärtused E count) saab leida võnkuva liikumise kvantiseerimise teel, mida peetakse ligikaudu harmooniliseks. Kõige lihtsamal kaheaatomilise molekuli puhul (üks vibratsiooniline vabadusaste, mis vastab tuumadevahelise kauguse muutusele r) peetakse harmooniliseks ostsillaator; selle kvantimine annab võrdse vahega energiatasemed:
E arv = h n e (u +1/2), (5)
kus n e on molekuli harmooniliste vibratsioonide põhisagedus, u on vibratsioonikvantarv, võttes väärtused 0, 1, 2, ... polüaatomilise molekuli iga elektroonilise oleku jaoks, mis koosneb N aatomid ( N³ 3) ja millel on f vibratsioonilised vabadusastmed ( f = 3N- 5 ja f = 3N- 6 vastavalt lineaarsete ja mittelineaarsete molekulide jaoks), selgub f nö normaalsed vibratsioonid sagedustega n i ( i = 1, 2, 3, ..., f) ja keerulist vibratsioonitasemete süsteemi:
![]()
Kus u i = 0, 1, 2, ... on vastavad vibratsioonikvantarvud. Tavaliste vibratsioonide sageduste kogum põhielektroonilises olekus on molekuli väga oluline omadus, olenevalt selle keemilisest struktuurist. Kõik või osa molekuli aatomitest osalevad teatud normaalses vibratsioonis; aatomid teostavad harmoonilisi vibratsioone sama sagedusega v i, kuid erineva amplituudiga, mis määravad vibratsiooni kuju. Tavalised vibratsioonid jagunevad kuju järgi venitamiseks (millel sidejoonte pikkused muutuvad) ja painutamiseks (millel muutuvad keemiliste sidemete vahelised nurgad - sidenurgad). Madala sümmeetriaga molekulide (ilma sümmeetriatelgedeta, mille suurusjärk on suurem kui 2) erinevate vibratsioonisageduste arv on võrdne 2-ga ja kõik vibratsioonid on mitte-mandunud ning sümmeetrilisemate molekulide puhul on kahe- ja kolmekordselt degenereerunud vibratsioonid (paarid ja kolmikud). sageduselt sobivate vibratsioonide kohta). Näiteks mittelineaarses kolmeaatomilises molekulis H2O f= 3 ja kolm mitte-mandunud vibratsiooni on võimalikud (kaks venitust ja üks painutamine). Sümmeetrilisemal lineaarsel kolmeaatomilisel CO 2 molekulil on f= 4 - kaks mitte-mandunud vibratsiooni (venitamine) ja üks kahekordselt degenereerunud (deformatsioon). Lameda ülisümmeetrilise molekuli C 6 H 6 puhul selgub f= 30 - kümme mitte-mandunud ja 10 topeltmandunud võnkumist; neist 14 vibratsiooni esineb molekuli tasapinnal (8 venitust ja 6 painutust) ja 6 tasandivälist painutusvibratsiooni - risti selle tasapinnaga. Veelgi sümmeetrilisemal tetraeedrilisel CH 4 molekulil on f = 9 - üks mitte-mandunud vibratsioon (venitus), üks kahekordselt degenereerunud (deformatsioon) ja kaks kolmekordselt degenereerunud (üks venitus ja üks deformatsioon).
Pöörlemisenergia tasemeid saab leida molekuli pöörlemisliikumise kvantiseerimise teel, käsitledes seda kindla ainega tahke ainena. inertsimomendid. Kõige lihtsamal kaheaatomilise või lineaarse polüaatomilise molekuli puhul selle pöörlemisenergia
![]()
Kus I on molekuli inertsimoment molekuli teljega risti oleva telje suhtes ja M- pöörlemismoment. Vastavalt kvantimisreeglitele,
![]()
kus on pöörlemiskvantarv J= 0, 1, 2, ... ja seega jaoks E rotatsioon vastu võetud:
kus pöörlemiskonstant määrab energiatasemete vahekauguste skaala, mis väheneb tuumamasside ja tuumadevaheliste kauguste kasvades.
Erinevat tüüpi M. s. tekivad erinevat tüüpi üleminekute käigus molekulide energiatasemete vahel. Vastavalt punktidele 1 ja 2
D E = E‘ - E'' = D E el + D E loe + D E pööra, (8)
kus muutused D E el, D E count ja D E elektrooniliste, vibratsiooni- ja pöörlemisenergiate pöörlemine vastab tingimusele:
D E el >> D E loe >> D E pööra (9)
[tasandite vahelised kaugused on samas järjekorras kui energiad ise E el, E ol ja E pöörlemine, rahuldav tingimus (4)].
D juures E el ¹ 0, saadakse elektrooniline mikroskoopia, mida vaadeldakse nähtavas ja ultraviolett- (UV) piirkonnas. Tavaliselt D E el ¹ 0 samaaegselt D E number 0 ja D E pöörlemine ¹ 0; erinev D E loe antud D jaoks E el vastavad erinevatele vibratsiooniribadele ja erinevatele D E pöörlemine antud D juures E el ja d E loendus - üksikud pöörlemisjooned, milleks see riba laguneb; saadakse iseloomulik triibuline struktuur.
![]()
N 2 molekuli elektron-vibratsiooniriba 3805 pöörlev poolitamine
Antud D-ga triipude komplekt E el (vastab puhtalt elektroonilisele üleminekule sagedusega v el = D E email/ h) nimetatakse ribasüsteemiks; üksikutel ribadel on erinev intensiivsus sõltuvalt üleminekute suhtelistest tõenäosustest, mida saab kvantmehaaniliste meetoditega ligikaudselt arvutada. Komplekssete molekulide puhul ühinevad ühe süsteemi ribad, mis vastavad antud elektroonilisele üleminekule, üheks laiaks pidevaks ribaks, mis võivad üksteisega kattuda. Orgaaniliste ühendite külmutatud lahustes täheldatud iseloomulikud diskreetsed elektroonilised spektrid . Elektroonilisi (täpsemalt elektron-vibratsiooni-pöörlemis-) spektreid uuritakse eksperimentaalselt, kasutades klaasist (nähtava piirkonna jaoks) ja kvartsist (UV-piirkonna jaoks) optikaga spektrograafe ja spektromeetreid, milles valguse lagundamiseks kasutatakse prismasid või difraktsioonvõresid. spekter .
D juures E el = 0 ja D E loetakse ¹ 0, saadakse võnkuvad magnetresonants, mida vaadeldakse lähedalt (kuni mitu µm) ja keskel (kuni mitukümmend µm) infrapuna (IR) piirkond, tavaliselt neeldumises, samuti valguse Ramani hajumises. Reeglina samaaegselt D E pöörlemine ¹ 0 ja etteantud juures E Tulemuseks on vibratsiooniriba, mis laguneb eraldi pöörlemisjoonteks. Need on kõige intensiivsemad võnkuvate M. s. triibud, mis vastavad D-le u = u’ - u'' = 1 (polüatomiliste molekulide puhul - D u i = u mina'- u i ''= 1 kohas D u k = u k' - u k '' = 0, kus k¹i).
Puhtalt harmooniliste vibratsioonide jaoks need valikureeglid, muude üleminekute keelamine toimub rangelt; anharmooniliste vibratsioonide korral ilmuvad ribad, mille puhul D u> 1 (ületoonid); nende intensiivsus on tavaliselt madal ja väheneb D suurenemisega u.
Vibratsiooni- (täpsemalt vibratsiooni-pöörlemis-) spektreid uuritakse eksperimentaalselt IR-piirkonnas neeldumisel, kasutades infrapunakiirgusele läbipaistvate prismadega või difraktsioonivõredega IR-spektromeetreid, samuti Fourier-spektromeetreid ja Ramani hajumist, kasutades suure avaga spektrograafe ( nähtav piirkond), kasutades laserergatust.
D juures E el = 0 ja D E arv = 0, saadakse puhtalt pöörlevad magnetsüsteemid, mis koosnevad üksikutest joontest. Nende imendumist täheldatakse kaugelt (sadu µm)IR piirkonnas ja eriti mikrolaine piirkonnas, samuti Ramani spektrites. Kahe- ja lineaarsete polüaatomiliste molekulide (nagu ka üsna sümmeetriliste mittelineaarsete polüaatomiliste molekulide) korral on need jooned (sagedusskaalal) üksteisest võrdsel kaugusel intervalliga Dn = 2 B neeldumisspektrites ja Dn = 4 B Ramani spektrites.
Puhtaid pöörlemisspektreid uuritakse neeldumises kaugemas IR piirkonnas, kasutades spetsiaalsete difraktsioonivõredega (ešelettidega) IR-spektromeetreid ja Fourier' spektromeetreid, mikrolainepiirkonnas mikrolaine (mikrolaine) spektromeetrite abil. , samuti Ramani hajumises suure avaga spektrograafide abil.
Mikroorganismide uurimisel põhinevad molekulaarspektroskoopia meetodid võimaldavad lahendada erinevaid keemia, bioloogia ja teiste teaduste probleeme (näiteks naftasaaduste, polümeersete ainete koostise määramine jne). Keemias MS järgi. uurida molekulide struktuuri. Elektrooniline M. s. võimaldavad saada teavet molekulide elektrooniliste kestade kohta, määrata ergastatud tasemeid ja nende omadusi ning leida molekulide dissotsiatsioonienergiaid (molekuli võnketasemete lähendamisel dissotsiatsioonipiiridele). Võnkuvate M. s. võimaldab leida iseloomulikke vibratsioonisagedusi, mis vastavad teatud tüüpi keemilistele sidemetele molekulis (näiteks lihtsad kaksik- ja kolmiksidemed C-C, C-H, N-H, O-H sidemed orgaaniliste molekulide puhul), erinevad aatomirühmad (näiteks CH 2 , CH 3 , NH 2), määrata molekulide ruumiline struktuur, eristada cis- ja trans-isomeere. Sel eesmärgil kasutatakse nii infrapuna neeldumisspektreid (IR) kui ka Ramani spektreid (RSS). IR-meetod on eriti laialt levinud kui üks tõhusamaid optilisi meetodeid molekulide struktuuri uurimiseks. See annab kõige täielikuma teabe koos SKR-meetodiga. Pöörlemismagnetresonantside, aga ka elektrooniliste ja vibratsioonispektrite pöörlemisstruktuuri uurimine võimaldab saada kogemusest leitud molekulide inertsmomentide väärtusi [mis saadakse pöörlemiskonstantide väärtustest, vt (7). )] leida suure täpsusega (lihtsamate molekulide, näiteks H 2 O puhul) molekuli tasakaalukonfiguratsiooni parameetrid - sideme pikkused ja sidemenurgad. Määratud parameetrite arvu suurendamiseks uuritakse isotoopmolekulide (eriti, milles vesinik on asendatud deuteeriumiga) spektreid, millel on samad tasakaalukonfiguratsiooni parameetrid, kuid erinevad inertsimomendid.
M. s. kasutamise näitena. Molekulide keemilise struktuuri määramiseks vaatleme benseeni molekuli C 6 H 6 . Tema M. s. kinnitab mudeli õigsust, mille kohaselt molekul on lame ja kõik 6 C-C sidet benseenitsüklis on samaväärsed ja moodustavad korrapärase kuusnurga, mille kuuendat järku sümmeetriatelg läbib molekuli sümmeetria keskpunkti risti temaga. lennuk. Elektrooniline M. s. neeldumisriba C 6 H 6 koosneb mitmest ribade süsteemist, mis vastavad üleminekutele maapealse paarissingli tasemelt ergastatud paaritutele tasemetele, millest esimene on kolmik ja kõrgemad on singletid. Triipude süsteem on kõige intensiivsem 1840. aasta piirkonnas ( E 5 - E 1 = 7,0 ev), on ribade süsteem kõige nõrgem 3400 ( E 2 - E 1 = 3,8ev), mis vastab singlett-tripleti üleminekule, mis on kogu spinni ligikaudsete valikureeglitega keelatud. Üleminekud vastavad ergastusele nn. p elektronid on delokaliseeritud kogu benseenitsüklis ; Elektrooniliste molekulaarspektritest saadud tasemediagramm on kooskõlas ligikaudsete kvantmehaaniliste arvutustega. Võnkuv M. s. C 6 H 6 vastavad sümmeetriakeskme olemasolule molekulis - IRS-is ilmuvad (aktiivsed) vibratsioonisagedused SRS-is puuduvad (mitteaktiivsed) ja vastupidi (nn alternatiivne keeld). 20 normaalsest C 6 H 6 vibratsioonist 4 on aktiivsed ICS-is ja 7 on aktiivsed SCR-is, ülejäänud 11 on passiivsed nii ICS-is kui ka SCR-is. Mõõdetud sageduse väärtused (in cm -1): 673, 1038, 1486, 3080 (ICS-is) ja 607, 850, 992, 1178, 1596, 3047, 3062 (TFR-is). Sagedused 673 ja 850 vastavad mittetasapinnalistele vibratsioonidele, kõik muud sagedused vastavad tasapinnalistele vibratsioonidele. Tasapinnalisele vibratsioonile on eriti iseloomulikud sagedus 992 (vastab C-C sidemete venitusvibratsioonile, mis koosneb benseenirõnga perioodilisest kokkusurumisest ja venitamisest), sagedused 3062 ja 3080 (vastavad C-H sidemete venitusvibratsioonile) ja sagedus 607. benseenirõnga painutusvibratsioonile). Vaadeldud C 6 H 6 võnkespektrid (ja sarnased C 6 D 6 võnkespektrid) on väga hästi kooskõlas teoreetiliste arvutustega, mis võimaldas anda nendest spektritest täieliku tõlgenduse ja leida kõigi normaalvibratsioonide kuju.
Samamoodi saate kasutada M. s. määrata erinevate klasside orgaaniliste ja anorgaaniliste molekulide, kuni väga keerukate, näiteks polümeeri molekulide struktuuri.
Loeng 12. Tuumafüüsika. Aatomituuma ehitus.
Tuum- see on aatomi keskne massiivne osa, mille ümber elektronid kvantorbiitidel tiirlevad. Tuuma mass on ligikaudu 4,10 3 korda suurem kui kõigi aatomis sisalduvate elektronide mass. Tuuma suurus on väga väike (10 -12 -10 -13 cm), mis on ligikaudu 105 korda väiksem kui kogu aatomi läbimõõt. Elektrilaeng on positiivne ja absoluutväärtuses võrdne aatomi elektronide laengute summaga (kuna aatom tervikuna on elektriliselt neutraalne).
Tuuma avastas E. Rutherford (1911) alfaosakeste hajumise katsetes ainet läbides. Olles avastanud, et a-osakesed hajuvad oodatust sagedamini suurte nurkade all, pakkus Rutherford, et aatomi positiivne laeng on koondunud väikesesse tuuma (enne seda valitsesid J. Thomsoni ideed, mille kohaselt aatomi positiivne laeng aatomit peeti ühtlaselt jaotunud kogu selle ruumala ulatuses) . Kaasaegsed Rutherfordi ideed kohe omaks ei võtnud (peamiseks takistuseks oli usk aatomielektronide paratamatusse langemisse tuumale, mis on tingitud tuuma ümber orbiidil liikudes elektromagnetkiirgusele kaovast energiast). Selle tunnustamisel mängis suurt rolli N. Bohri kuulus töö (1913), mis pani aluse aatomi kvantteooriale. Bohr postuleeris orbiitide stabiilsust kui aatomi elektronide liikumise kvantimise algprintsiipi ja tuletas sellest seejärel joonoptiliste spektrite seadused, mis selgitasid ulatuslikku empiirilist materjali (Balmeri seeria jne). Mõnevõrra hiljem (1913. aasta lõpus) näitas Rutherfordi õpilane G. Moseley eksperimentaalselt, et aatomite joonröntgenispektri lühilainepiiri nihe elemendi aatomarvu Z muutumisel perioodilises elementide süsteemis vastab Bohri teooriale, kui eeldada, et tuuma elektrilaeng (elektronilaengu ühikutes) on võrdne Z. See avastus purustas täielikult usaldamatuse barjääri: uus füüsiline objekt - tuum - osutus kindlalt ühendatud terve hulga pealtnäha heterogeensete nähtustega, mis on nüüdseks saanud ühtse ja füüsiliselt läbipaistva seletuse. Pärast Moseley tööd tehti füüsikas lõplikult kindlaks aatomituuma olemasolu fakt.
Kerneli koostis. Tuuma avastamise ajal oli teada vaid kaks elementaarosakest – prooton ja elektron. Sellest lähtuvalt peeti tõenäoliseks, et tuum neist koosneb. Siiski 20ndate lõpus. 20. sajandil Prooton-elektron-hüpoteesil tekkis tõsine raskus, mida nimetatakse lämmastikukatastroofiks: prooton-elektroni hüpoteesi kohaselt peaks lämmastiku tuum sisaldama 21 osakest (14 prootonit ja 7 elektroni), millest igaühe spinn oli 1/2. . Lämmastiku tuuma spinn oleks pidanud olema pooltäisarv, kuid optiliste molekulaarspektrite mõõtmise andmetel osutus spinn võrdseks 1-ga.
Tuuma koostis selgitati pärast J. Chadwicki avastamist (1932) neutron. Neutroni mass, nagu selgus juba Chadwicki esimestest katsetest, on lähedane prootoni massile ja spinn on võrdne 1/2-ga (määratud hiljem). Idee, et tuum koosneb prootonitest ja neutronitest, väljendas esmakordselt trükis D. D. Ivanenko (1932) ja kohe pärast seda arendas välja W. Heisenberg (1932). Eeldus tuuma prooton-neutron koostise kohta leidis hiljem eksperimentaalselt täielikult kinnitust. Kaasaegses tuumafüüsikas kombineeritakse prootonit (p) ja neutronit (n) sageli üldnimetuse nukleon all. Nukleonide koguarvu tuumas nimetatakse massiarvuks A, prootonite arv võrdub tuuma laenguga Z (elektronide laengu ühikutes), neutronite arv N = A–Z. U isotoobid sama Z, aga erinev A Ja N, on tuumadel samad isobaarid A ja erinevad Z ja N.
Seoses uute nukleonitest raskemate osakeste avastamisega tekkisid nn. nukleoni isobaarid, selgus, et ka nemad peaksid olema osa tuumast (intranukleaarsed nukleonid, põrkudes üksteisega, võivad muutuda nukleoni isobaarideks). Kõige lihtsamas tuumas - deuteron , mis koosneb ühest prootonist ja ühest neutronist, peaksid nukleonid jääma nukleoni isobaaride kujul ~ 1% ajast. Mitmed täheldatud nähtused annavad tunnistust selliste isobaariliste olekute olemasolust tuumades. Lisaks nukleonidele ja nukleoni isobaaridele ka tuumades perioodiliselt lühiajaliselt (10 -23 -10 -24 sek) ilmuvad mesonid , sealhulgas kergeim neist - p-mesonid. Nukleonide interaktsioon taandub mesoni mitmekordsele emissioonile ühe nukleoni poolt ja selle neeldumisele teise poolt. Tekkiv st. vahetusmesoni voolud mõjutavad eelkõige tuumade elektromagnetilisi omadusi. Mesonivahetusvoolude kõige selgem ilming leiti deuteroni lõhenemise reaktsioonis suure energiaga elektronide ja g-kvantide poolt.
Nukleonide interaktsioon. Jõud, mis hoiavad tuumas nukleone, nimetatakse tuumaenergia . Need on kõige tugevamad füüsikas teadaolevad vastasmõjud. Tuumas kahe nukleoni vahel mõjuvad tuumajõud on suurusjärgus sada korda intensiivsemad kui prootonite elektrostaatiline interaktsioon. Tuumajõudude oluline omadus on nende. sõltumatus nukleonite laenguseisundist: kahe prootoni, kahe neutroni või neutroni ja prootoni tuuma vastasmõju on sama, kui nende osakeste paaride suhtelise liikumise olekud on samad. Tuumajõudude suurus sõltub nukleonide vahelisest kaugusest, nende spinnide vastastikusest orientatsioonist, spinnide orientatsioonist orbiidi nurkimpulsi ja ühest osakesest teise tõmmatud raadiuse vektori suhtes. Tuumajõude iseloomustab teatav toimeulatus: nende jõudude potentsiaal väheneb koos kaugusega r osakeste vahel kiiremini kui r-2, ja jõud ise on kiiremad kui r-3. Tuumajõudude füüsikalise olemuse arvestamisest järeldub, et need peaksid kaugusega plahvatuslikult vähenema. Tuumajõudude toimeraadiuse määrab nn. Comptoni lainepikkus r 0 mesoneid, mis interaktsiooni ajal nukleonide vahel vahetatakse:
siin m on mesonimass, on Plancki konstant, Koos- valguse kiirus vaakumis. P-mesonite vahetusest põhjustatud jõud on suurima toimeraadiusega. Nende jaoks r 0 = 1,41 f (1 f = 10 -13 cm). Tuumadevahelised kaugused tuumades on täpselt selles suurusjärgus, kuid tuumajõududele aitavad kaasa ka raskemate mesonite (m-, r-, w-mesonid jne) vahetused. Kahe nukleoni vaheliste tuumajõudude täpset sõltuvust kaugusest ja tuumajõudude panusest erinevat tüüpi mesonite vahetusest ei ole kindlalt kindlaks tehtud. Mitmetuumalistes tuumades on võimalikud jõud, mida ei saa taandada ainult nukleonipaaride vastastikmõjule. Rolli nende nn Paljude osakeste jõud tuumade struktuuris jääb ebaselgeks.
Kerneli suurused sõltuvad neis sisalduvate nukleonide arvust. Tuumas olevate nukleonide arvu p keskmine tihedus (nende arv ruumalaühiku kohta) kõigi mitmetuumaliste tuumade puhul (A > 0) on peaaegu sama. See tähendab, et tuuma maht on võrdeline nukleonide arvuga A ja selle lineaarne suurus ~A 1/3. Efektiivne südamiku raadius R määratakse seosega:
R = a A 1/3 , (2)
kus on konstant A lähedal Hz, kuid erineb sellest ja sõltub sellest, millistes füüsikalistes nähtustes seda mõõdetakse R. Tuuma nn laenguraadiuse korral mõõdetuna elektronide hajumise järgi tuumadel või energiatasemete asendi järgi m- mesoaatomid : a = 1,12 f. Interaktsiooniprotsesside põhjal määratud efektiivne raadius hadronid (nukleonid, mesonid, a-osakesed jne), mille tuumad on laengust veidi suuremad: alates 1,2 f kuni 1,4 f.
Tuumaaine tihedus on tavaliste ainete tihedusega võrreldes fantastiliselt kõrge: see on ligikaudu 10 14 G/cm 3. Tuumas on r keskosas peaaegu konstantne ja perifeeria suunas väheneb eksponentsiaalselt. Empiiriliste andmete ligikaudseks kirjeldamiseks aktsepteeritakse mõnikord järgmist r-i sõltuvust kaugusest r tuuma keskpunktist:
![]() .
.
Efektiivne südamiku raadius R võrdne R 0 + b. Väärtus b iseloomustab tuuma piiri hägusust, see on peaaegu sama kõigi tuumade puhul (» 0,5 f). Parameeter r 0 on kahekordne tihedus tuuma "piiril", mis on määratud normaliseerimistingimusest (p mahuintegraali võrdsus nukleonide arvuga A). (2) järeldub, et tuumade suurused varieeruvad suurusjärgus 10-13 cm kuni 10-12 cm raskete tuumade jaoks (aatomi suurus ~ 10 -8 cm). Valem (2) kirjeldab aga tuumade lineaarsete mõõtmete suurenemist nukleonide arvu suurenemise korral ainult umbkaudselt, märkimisväärse suurenemisega A. Tuuma suuruse muutumine ühe või kahe nukleoni lisandumisel sellele sõltub tuuma ehituse detailidest ja võib olla ebaregulaarne. Eelkõige (nagu näitavad aatomi energiatasemete isotoopnihke mõõtmised) mõnikord tuuma raadius isegi väheneb, kui lisada kaks neutronit.
MOLEKULAARSPEKTRID
Vabadele või nõrgalt seotud molekulidele kuuluva valguse emissiooni-, neeldumis- ja Ramani spektrid. Tüüpilised mikroskoopilised süsteemid on triibulised, neid vaadeldakse enam-vähem kitsaste ribade kujul spektri UV-, nähtava- ja IR-piirkondades; piisava spektriseadmete eraldusvõimega mol. triibud lagunevad tihedalt asetsevate joonte kogumiks. M. s. struktuur. erinev erinevate jaoks molekule ja muutub keerulisemaks, kui aatomite arv molekulis suureneb. Väga keeruliste molekulide nähtav ja UV-spekter on üksteisega sarnased ja koosnevad mõnest laiast pidevast ribast. M. s. tekivad energiatasemete vaheliste kvantüleminekute ajal?" ja?" molekulid vastavalt suhtele:
kus hv on sagedusega v emiteeritud või neeldunud footoni energia. Ramani hajumises on hv võrdne langevate ja hajutatud footonite energiate erinevusega. M. s. palju keerulisem kui aatomispektrid, mille määrab sisemise suurem keerukus liikumised molekulis, sest lisaks elektronide liikumisele kahe või enama tuuma suhtes toimub molekulis võnkumine. tuumade liikumine (koos neid ümbritsevate siseelektronidega) ümber tasakaaluasendi ja pöörlevad. selle liikumist tervikuna. Elektrooniline, võnkuv ja pöörake. Molekuli liikumised vastavad kolmele energiatasemele el, ?col ja?vr ning kolmele tüübile M. s.
Vastavalt kvant. mehaanika, igat tüüpi liikumise energia molekulis võib omandada ainult teatud väärtused (kvanteeritud). Molekuli koguenergia? võib ligikaudu esitada kvantiseeritud energia väärtuste summana, mis vastab kolmele siseenergia tüübile. liigutused:
??el +?col+?vr, (2) ja suurusjärgus
El: col: vr = 1: m/M: m/M, (3)
kus m on elektroni mass ja M on suurusjärgus molekulis olevate aatomite tuumade massist, s.o.
El -> ?count ->?vr. (4) Tavaliselt el tellida mitu. eV (sadu kJ/mol), ?col = 10-2-10-1 eV, ?vr=10-5-10-3 eV.
Molekuli energiatasemete süsteemi iseloomustavad üksteisest kaugel asuvate elektrooniliste energiatasemete komplektid (disag. ?el at?col=?time=0). vibratsioonitasemed, mis asuvad üksteisele palju lähemal (antud el ja aja diferentsiaalväärtused = 0) ja veelgi lähemal üksteisele pöörlemistasemed (aja väärtused antud el ja aja jaoks).
Elektroonilise energia tasemed a kuni b joonisel fig. 1 vastavad molekuli tasakaalukonfiguratsioonidele. Iga elektrooniline olek vastab teatud tasakaalukonfiguratsioonile ja teatud väärtusele?el; väikseim väärtus vastab põhiväärtusele. elektrooniline olek (molekuli elektrooniline põhienergia tase).
Riis. 1. Kaheaatomilise molekuli energiatasemete diagramm, a ja b - elektroonilised tasemed; v" ja v" on kvant. võnkumiste arv tasemed; J" ja J" - kvant. numbreid pööratakse. tasemed.
Molekuli elektrooniliste olekute komplekti määravad selle elektroonilise kesta omadused. Põhimõtteliselt saab ?el väärtusi arvutada kvantmeetoditega. keemias aga saab seda probleemi lahendada vaid ligikaudselt ja suhteliselt lihtsate molekulide puhul. Oluline teave molekulide elektrooniliste tasemete kohta (nende asukoht ja omadused), mis on määratud selle keemilise ainega. struktuur saadakse uurides M. s.
Elektroonilise energiataseme väga oluline tunnus on kvantarvu 5 väärtus, mis määrab abs. kõigi elektronide kogupöörlemismomendi väärtus. Keemiliselt stabiilsetel molekulidel on reeglina paarisarv elektrone ja nende jaoks 5 = 0, 1, 2, . . .; peamiseks elektrooniline tase on tavaliselt 5=0, ergastatud tasemete puhul - 5 = 0 ja 5=1. Nimetatakse tasemeid, kus S=0. singlett, S=1 - triplett (kuna nende kordsus on c=2S+1=3).
Kahe- ja lineaarsete kolmeaatomiliste molekulide puhul iseloomustatakse elektroonilisi tasemeid kvantväärtustega. arv L, mis määrab abs. kõigi elektronide kogu orbiidi impulsi projektsiooni suurus molekuli teljele. Taseme L=0, 1, 2, ... tähistatakse vastavalt S, P, D. . ., ja ja seda tähistab vasakus ülanurgas olev indeks (näiteks 3S, 2P). Sümmeetriakeskmega molekulide puhul (nt CO2, CH6) jagatakse kõik elektroonilised tasemed paaris- ja paarituteks (vastavalt g ja u) olenevalt sellest, kas neid defineeriv lainefunktsioon säilitab oma märgi, kui see on ümberpööratud. sümmeetria keskpunkt.
Vibratsioonienergia tasemeid saab leida vibratsiooni kvantifitseerimise teel. liigutused, mida peetakse ligikaudu harmoonilisteks. Kaheaatomilist molekuli (üks võnkevabadusaste, mis vastab tuumadevahelise kauguse r muutumisele) võib pidada harmooniliseks. ostsillaator, mille kvantimine annab võrdse vahega energiatasemed:
kus v - peamine. harmooniline sagedus molekuli vibratsioonid, v=0, 1, 2, . . .- võnkuma kvant. number.
3 aatomist koosneva polüaatomilise molekuli iga elektroonilise oleku jaoks, millel on f võnkumine. vabadusastmed (lineaarsete ja mittelineaarsete molekulide puhul vastavalt f=3N-5 ja f=3N-6), selgub / nn. normaalvõnkumised sagedustega vi(ill, 2, 3, ..., f) ja kompleksne võnkesüsteem. energiatasemed:
Sageduste komplekt on normaalne. kõikumised põhiliselt nähtuste elektrooniline seisund. molekuli oluline omadus, olenevalt selle keemilisest koostisest. hooned. Teatud mõttes. vibratsioonid hõlmavad kas kõiki molekuli aatomeid või osa neist; aatomid täidavad harmoonilisi võnkumised sama sagedusega vi, kuid erinevaga amplituudid, mis määravad vibratsiooni kuju. Tavaline võnked jagunevad kuju järgi valentsiks (keemiliste sidemete pikkused muutuvad) ja deformatsiooniks (muutuvad keemiliste sidemete vahelised nurgad - sidenurgad). Madalama sümmeetriaga molekulide puhul (vt MOLEKULI SYMMETRIA) f=2 ja kõik vibratsioonid ei ole degenereerunud; sümmeetrilisemate molekulide jaoks on kahe- ja kolmekordselt degenereerunud vibratsioonid, st sageduselt ühtivad vibratsioonipaarid ja kolmikud.
Pöörlemisenergia tasemeid saab leida pöörlemist kvantifitseerides. molekuli liikumine, pidades seda televiisoriks. teatud inertsimomentidega keha. Kahe- või lineaarse kolmeaatomilise molekuli puhul on selle pöörlemisenergia? liikumise kvantiteedi hetk. Vastavalt kvantimisreeglitele,
M2=(h/4pi2)J(J+1),
kus f=0, 1,2,. . .- pöörlemiskvant. number; jaoks saame:
Вр=(h2/8pi2I)J(J+1) = hBJ(J+1), (7)
kus nad pöörlevad. konstant B=(h/8piI2)I
määrab energiatasemete vahekauguste skaala, mis väheneb tuumamasside ja tuumadevaheliste kauguste kasvades.
Diff. tüübid M. s. tekivad siis, kui erinevad molekulide energiatasemete vahelise ülemineku tüübid. Vastavalt punktidele 1 ja 2:
D?=?"-?"==D?el+D?col+D?vr,
ja sarnaselt (4) D?el->D?count->D?time. D'el00 juures saadakse elektrooniline mikroskoopia, mis on jälgitav nähtavas ja UV-piirkonnas. Tavaliselt D100 juures nii D'number 0 kui ka D' time 0; diff. D? count antud D el vastama diff. võnkuma triibud (joon. 2) ja lagunemine. D?vr antud D?el ja D?arv dep. pöörata jooned, milleks võnkumised lagunevad. triibud (joonis 3).

Riis. 2. Elektroino-võnkumine. N2 molekuli spekter lähi-UV-piirkonnas; triipude rühmad vastavad diff. väärtused Dv= v"-v".
Nimetatakse antud D?el-iga sagedusribade komplekt (mis vastab puhtelektroonilisele üleminekule sagedusega nel=D?el/h). ribasüsteem; triibud on erinevad intensiivsus sõltuvalt suhtelisest ülemineku tõenäosused (vt KVANTÜLEMINEK).

Riis. 3. Pöörake. elektron-kolsbati lõhenemine. triibud 3805.0 ? N2 molekulid.
Komplekssete molekulide puhul ühinevad ühe süsteemi ribad, mis vastavad antud elektroonilisele üleminekule, tavaliselt üheks laiaks pidevaks ribaks; võivad kattuda üksteisega ja mitu korda. sellised triibud. Külmutatud orgaanilistes lahustes täheldatakse iseloomulikke diskreetseid elektroonilisi spektreid. ühendused.
Elektroonilisi (täpsemalt elektron-vibratsiooni-pöörlemis-) spektreid uuritakse klaasi (nähtav piirkond) ja kvartsist (UV-piirkond, (vt UV-KIIRGUS)) optikaga spektriinstrumentidega. Kui D2el = 0 ja Docol 0, saadakse võnkumised. Lähis-IR piirkonnas täheldatud MS on tavaliselt neeldumis- ja Ramani spektris. Reeglina antud D arvu ja võnke? riba laguneb osadeks. pöörata read. Kõige intensiivsem vibratsiooni ajal. M. s. ribad, mis vastavad tingimusele Dv=v"- v"=1 (polüatomiliste molekulide puhul Dvi=v"i- v"i=1 Dvk=V"k-V"k=0; siin määravad i ja k erinevad normaalvibratsioonid). Puhtalt harmooniliseks kõikumised, järgitakse neid valikureegleid rangelt; anharmooniliste jaoks vibratsiooni jaoks ilmuvad ribad, mille puhul Dv>1 (ületoonid); nende intensiivsus on tavaliselt madal ja väheneb Dv suurenemisega. Võnkumine M. s. (täpsemalt vibratsiooni-rotatsiooni) uuritakse IR spektromeetrite ja Fourier spektromeetrite abil ning Ramani spektreid uuritakse suure avaga spektrograafidega (nähtava piirkonna jaoks), kasutades laserergatust. Kui D2el=0 ja Docol=0, saadakse puhas pöörlemine. spektrid, mis koosnevad eraldi read. Neid täheldatakse neeldumisspektrites kaugemas IR piirkonnas ja eriti mikrolaine piirkonnas, samuti Ramani spektrites. Kaheaatomiliste, lineaarsete kolmeaatomiliste molekulide ja üsna sümmeetriliste mittelineaarsete molekulide puhul on need jooned (sagedusskaalal) üksteisest võrdsel kaugusel.
Pöörake puhtalt. M. s. uuriti IR-spektromeetrite abil spetsiaalsete difraktsioon võred (ešeletid), Fourier spektromeetrid, tagurpidi laine lambil põhinevad spektromeetrid, mikrolaine (mikrolaine) spektromeetrid (vt SUBMILLIMETERSPEKTROSKOPIA, MIKROLAINE SPEKTROSKOPIA) ja pöörlevad. Ramani spektrid – suure avaga spektromeetrite kasutamine.
Molekulaarspektroskoopia meetodid, mis põhinevad mikroskoopia uurimisel, võimaldavad lahendada erinevaid keemiaprobleeme. Elektrooniline M. s. anda teavet elektrooniliste kestade, ergastatud energiatasemete ja nende omaduste kohta, molekulide dissotsiatsioonienergia kohta (energiatasemete lähenemisel dissotsiatsioonipiirile). Võnkumiste uurimine. spektrid võimaldavad leida iseloomulikke vibratsioonisagedusi, mis vastavad teatud tüüpi kemikaalide olemasolule molekulis. sidemed (näiteks kaksik- ja kolmiksidemed C-C, C-H, N-H sidemed orgaaniliste molekulide puhul), määravad ruumid. struktuuri, eristada cis- ja trans-isomeere (vt Molekulide ISOMEERIStika). Eriti laialt levinud on infrapunaspektroskoopia meetodid – üks tõhusamaid optilisi meetodeid. Molekulide struktuuri uurimise meetodid. Need pakuvad kõige täielikumat teavet koos Ramani spektroskoopia meetoditega. Uuring hakkab pöörlema. spektreid ja ka pöörlema. elektroonika- ja vibratsioonistruktuurid. M. s. võimaldab kasutada eksperimentaalselt leitud molekulide inertsimomente, et leida suure täpsusega tasakaalukonfiguratsioonide parameetreid – sidemepikkusi ja sidenurki. Määratud parameetrite arvu suurendamiseks uuritakse isotoopspektreid. molekulid (eelkõige molekulid, milles vesinik on asendatud deuteeriumiga), millel on samad tasakaalukonfiguratsiooni parameetrid, kuid erinevad. inertsimomendid.
M. s. Neid kasutatakse ka spektraalanalüüsis aine koostise määramiseks.
- - kristallid, mis on moodustunud molekulidest, mis on omavahel seotud nõrkade van der Waalsi jõudude või vesiniksidemetega...
Füüsiline entsüklopeedia
- - kvantkeemias integraalavaldiste nimetused, mida kasutatakse maatriksisse kirjutamiseks, moodustavad elektroonilise Schrödingeri võrrandi, mis määrab mitmeelektronilise molekuli elektronlaine funktsioonid...
Keemia entsüklopeedia
- - moodustuvad formaalselt valents-küllastusest. Molekulid molekulidevahelise interaktsiooni jõudude tõttu...
Keemia entsüklopeedia
- - moodustuvad van der Waalsi jõududega seotud molekulidest. Molekulide sees on aatomid omavahel seotud palju tugevamate sidemetega...
Keemia entsüklopeedia
- - org-molekulide visuaalne esitus. ja mitte-org. ühendid, mis võimaldavad hinnata molekulis sisalduvate aatomite suhtelist asukohta...
Keemia entsüklopeedia
- - elektromagnetilise emissiooni ja neeldumise spektrid. kiirgus ja kombinatsioon ...
Keemia entsüklopeedia
- - Vt Osaliselt seotud...
- - molekulide vastastikmõju jõud, mis olenevalt välistingimustest määravad ühe või teise aine agregatsiooni oleku ja hulga muid füüsikalisi omadusi...
Hüdrogeoloogia ja insenerigeoloogia sõnastik
- - valguse optilise neeldumise, emissiooni ja Ramani hajumise spektrid, mis tekivad molekulide üleminekul ühelt energiatasemelt teisele. M. s. koosnevad enam-vähem laiadest triipudest, piltidest...
Suur entsüklopeediline polütehniline sõnaraamat
- - Artiklid täiturbioloogilised mootoridbioloogilised nanoobjektid biomeditsiinilised mikroelektromehaanilised süsteemid biopolümeeridravimite kohaletoimetamise kiibid kiibi multifunktsionaalsete nanoosakeste laboris...
Nanotehnoloogia entsüklopeediline sõnastik
- - optiline vabadele või nõrgalt seotud molekulidele kuuluva valguse emissiooni, neeldumise ja hajumise spektrid...
Loodusteadus. Entsüklopeediline sõnaraamat
- - kaasasündinud ainevahetushäired, pärilikest ainevahetushäiretest põhjustatud haigused. Mõiste "M. b." pakkus välja Ameerika keemik L. Pauling...
- - kristallid, mis on moodustunud molekulidest, mis on omavahel seotud nõrkade van der Waalsi jõudude või vesiniksidemetega. Molekulide sees toimivad aatomite vahel tugevamad kovalentsed sidemed...
Suur Nõukogude entsüklopeedia
- - emissiooni ja neeldumise optilised spektrid, samuti valguse Ramani hajumine, mis kuuluvad vabade või nõrgalt ühendatud molekulide hulka. M. s. on keerulise struktuuriga...
Suur Nõukogude entsüklopeedia
- - vabadele või nõrgalt seotud molekulidele kuuluva valguse emissiooni, neeldumise ja hajumise optilised spektrid...
Suur entsüklopeediline sõnastik
- - või osalised tegevused...